Bei Markteinführung war das Gen-Therapeutikum Zolgensma® als „teuerstes Medikament der Welt“ umstritten. Für Kinder, die unter Spinaler Muskelatrophie leiden, ist der medizinische Nutzen allerdings erheblich. „Vor 20 Jahren ist die Mehrheit der Kinder mit SMA nach dem ersten Lebensjahr verstorben“, sagt Univ.-Prof. Heymut Omran, Direktor der Klinik für Kinder- und Jugendmedizin am UKM (Universitätsklinikum Münster). Ab dem Sommer wird der Test auf SMA Bestandteil des Neugeborenen-Screenings und kann so frühestmöglich diagnostiziert und therapiert werden. Erstmals ist die Heilung der eigentlich todbringenden Erkrankung möglich.
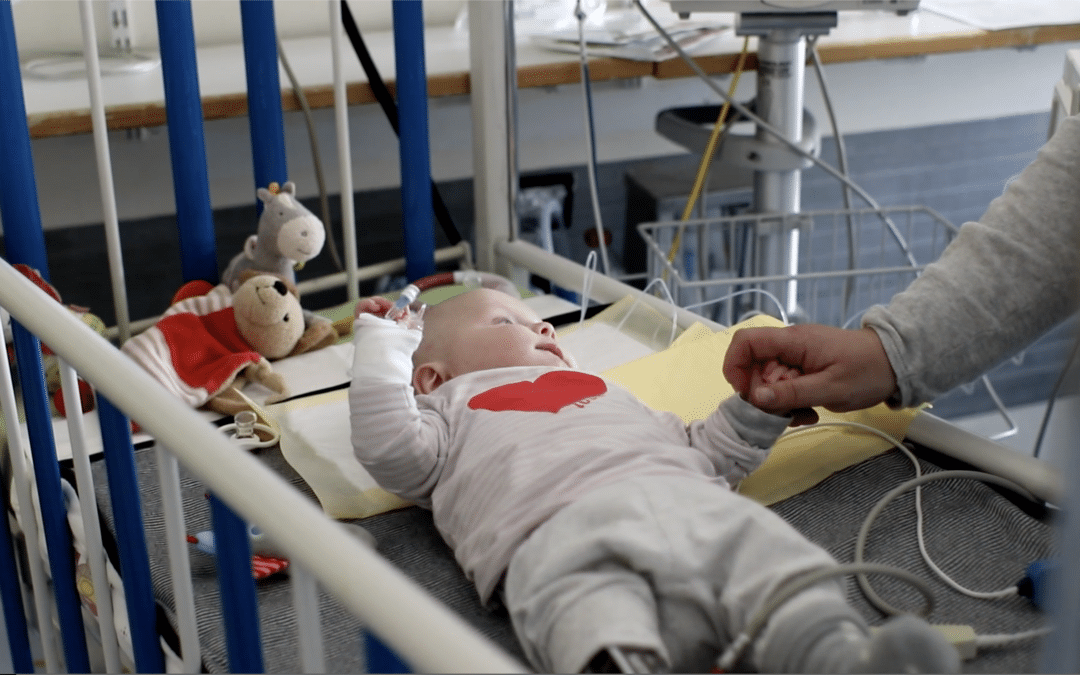
Münster (ukm/aw) – Amy L. hat großes Glück: Das vier Monate alte Baby bekommt heute eine Infusion, die sein Leben voraussichtlich retten und verbessern wird. Amy wurde schon als Neugeborenes im Rahmen eines Modellprojekts auf SMA gescreent. Direkt nach der Geburt stellte sich so heraus, dass Amy von der erblichen Motoneuronenerkrankung betroffen ist. Ein Schock für ihre Eltern, die schon drei gesunde Kinder haben. Bei der SMA Typ 1 ist die Rumpfnahe Muskulatur von zunehmender Schwäche beeinträchtigt – Drehen, Robben und Sitzen werden gar nicht erst erlernt. „Bei diesen schweren Verlaufsformen sind die Kinder hypoton und haben zunehmend weniger Kraft in den Rumpfnahen Muskeln, können zum Beispiel den Kopf in Bauchlage nicht heben“, so Kinderneurologe Dr. Oliver Schwartz, der am UKM derzeit rund 40 Kinder mit SMA betreut.
Bei fortschreitender Erkrankung ist auch die Atemmuskulatur betroffen – die Kinder verstarben früher unbehandelt häufig innerhalb der ersten zwei Lebensjahre. „Erst seit wenigen Jahren stehen uns überhaupt kurative Therapieoptionen zur Verfügung, um diesen Kindern zu helfen“, sagt der Kinderneurologe und Kinderpneumologe Univ.-Prof. Heymut Omran. Zunächst handelte es sich um Spinraza®. Seit 2020 steht Zolgensma® zur Verfügung, das anders als das Vorgängertherapeutikum nicht über eine Lumbalpunktion alle vier Monate, sondern als Einmalgabe intravenös verabreicht werden kann. Wie aber wirkt das Medikament, das Schwartz bezeichnender Weise als „Genersatztherapeutikum“ bezeichnet? Bei der SMA ist das Gen SMN1 defekt. Zolgensma® schleust mittels eines Virus, das als „Genfähre“ fungiert, eine gesunde Kopie des SMN1-Gens in die Körperzellen. Die Folge: Die Muskelatrophie setzt nicht ein – prognostisch ist den Kindern ein Leben ohne Beeinträchtigung möglich.
Amy bekommt an diesem Morgen die rettende Injektion: Über einen Venenzugang am Kopf wird das Medikament ihrem kleinen Körper innerhalb von 60 Minuten zugeführt. Zuvor wurde die Spritze mit Zolgensma® in der UKM-Apotheke steril aufbereitet und an Schwartz persönlich über eine Sicherheitsschleuse übergeben. Eine Maßnahme, die dem hohen Preis des Therapeutikums geschuldet ist: Die Einmaldosis Zolgensma® kostet rund 2 Millionen Euro. Amy weiß von all dem nichts und lässt alles geduldig geschehen. Für Klinikdirektor Omran grenzt die neue Therapiemöglichkeit bei SMA nach eigener Aussagen „an ein Wunder“. So können die Mediziner sogar bei einigen Kindern, bei denen die Muskelatrophie bereits eingesetzt hatte, beobachten, dass sie die Kopfkontrolle wiedererlangen und sich die Bewegungsabläufe normalisieren.
Er habe in den 20 Jahren seiner klinischen und wissenschaftlichen Tätigkeit viele Kinder mit SMA aufgeben müssen, die meist innerhalb der ersten Lebensjahre starben, berichtet der Kinder- und Jugendmediziner. Mit dem SMA-Neugeborenen-Screening, für dessen deutschlandweite Einführung ab diesem Sommer er als pädiatrisches Mitglied der Gendiagnostik-Kommission des Robert-Koch-Instituts (RKI) mitverantwortlich zeichnet, hat sich die Situation der Kinder dramatisch verbessert. „Wenn man die spinale Muskelatrophie so früh wie möglich diagnostiziert, kann man diesen Kindern so helfen, dass sie gar nicht krank werden. Das ist etwas, was wir am liebsten haben – so ähnlich wie bei einer Impfung – dass wir das Ausbrechen einer Erkrankung verhüten können“, sagt Omran.
Für weiterführende Genersatztherapien bei anderen Gendefekten sei zumindest eine Basis geschaffen, auf deren Grundlage geforscht werden könne. „Überall dort, wo wir von etwas ‚zu wenig‘ haben, könnte das Modell des Ersetzens dieses fehlenden Stoffs im Erbgut ein Ansatz sein. Daran müssen wir weiter forschen. Wir sehen, dass wir in der Lage sind das Genom zu therapieren. Gleichzeitig können wir durch das Neugeborenen-Screening Kinder frühzeitig diagnostizieren, einer Therapie zuführen und so retten. Das macht unser kinderneurologisches Team am UKM sehr froh.“ (Ein ausführliches Experteninterview zur Entwicklung der Behandlungsoptionen von SMA finden Sie hier.)






 Der Notfalldienst ist über die kostenfreie Rufnummer:
Der Notfalldienst ist über die kostenfreie Rufnummer: